El Síndrome de Dravet, también conocido como Epilepsia Mioclónica Severa de la Infancia (SMEI), fue descrito en 1978 por la psiquiatra y epileptóloga Charlotte Dravet. Desde 1985 la Liga Internacional contra la Epilepsia (ILAE) lo incluye dentro del apartado de ‘Epilepsias y síndromes indeterminados respecto a la localización con crisis generalizadas y focales’.
Se trata de una encefalopatía epiléptica que se caracteriza por crisis epilépticas frecuentes con convulsiones febriles y afebriles generalizadas y/o parciales recurrentes y prolongadas, con una edad de aparición entre 4 y 12 meses y refractaria a tratamiento. En edades más avanzadas, las convulsiones pueden evolucionar a mioclonías, ausencias atípicas y convulsiones asociadas a retraso psicomotor y ataxias. El desarrollo cognitivo se vuelve progresivamente más pobre a partir del segundo año de edad, derivando en retraso cognitivo en la mayoría de los casos, de grado moderado a severo.
El Síndrome de Dravet es una enfermedad de origen genético y se encuadra dentro de la familia patológica de canalopatías, ya que aproximadamente el 75% de los pacientes afectados presenta una mutación en el gen SCN1A.
Esta enfermedad es una de las variantes más severa de las canalopatías descritas hasta la fecha. Las convulsiones febriles, una variante benigna de la enfermedad, también forman parte de esta familia.
Epilepsias relacionadas
Grupo de epilepsias relacionadas con una causa genética similar (de menor a mayor severidad):
¿Cuántos afectados existen?
El Síndrome de Dravet no se describió hasta finales de 1970 y hasta 2003 no existió un test genético que ayudara a diagnosticar la enfermedad. Esto explica que el número de afectados no se conozca con exactitud. Se estima que la incidencia de la enfermedad es de 1 entre 20.000 nacimientos, lo que la encuadra en el grupo de enfermedades raras (1/2.500).
Aproximadamente un 25% de los pacientes presenta una historia familiar de epilepsia o convulsiones febriles de diferente grado. Según los cálculos de la Fundación Síndrome de Dravet, en España debe haber entre 200 y 400 niños con este problema. Sin embargo, el número puede ser mucho mayor, ya que el diagnóstico clínico en ocasiones resulta complicado.
|
Síntomas clínicos
El Síndrome de Dravet es un trastorno del neurodesarrollo que se caracteriza por una epilepsia severa resistente al tratamiento que presenta las siguientes características clínicas y electroencefalográficas:
Inicio en el primer año de vida
Desarrollo cognitivo normal previo al inicio de la crisis
Resistencia al tratamiento farmacológico
Crisis convulsivas prolongadas (más de diez minutos)
Normalidad inicial del EEG con deterioro posterior, asociado a deterioro cognitivo progresivo con ataxia y otras alteraciones motoras
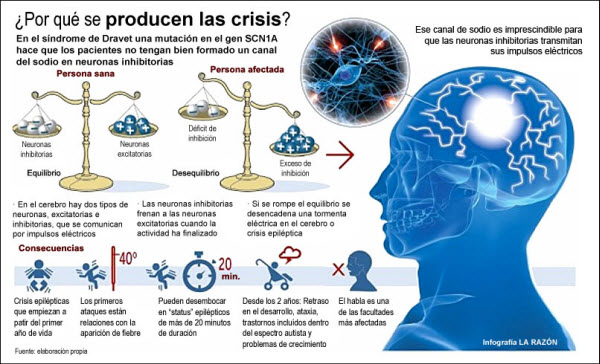
En la mayoría de los casos, las crisis epilépticas comienzan a partir del primer año de vida. Las primeras crisis están relacionadas con la aparición de fiebre y son convulsiones generalizadas tónicas-clónicas o unilaterales. En muchas ocasiones estas crisis desembocan en status epilepticus, episodios de más de veinte minutos de duración. Con el tiempo, también aparecen crisis afebriles o relacionadas con otros estímulos, y otro tipo de convulsiones como las mioclonías (sacudidas musculares breves), ausencias atípicas y crisis parciales-complejas.
A partir del segundo año se empiezan a observar síntomas de retraso en el desarrollo cognitivo y psicomotor. En muchos casos se observan ataxia, trastornos incluidos dentro del espectro autista, problemas alimenticios, de crecimiento y trastornos del sueño. El habla suele ser una de las facultades más afectadas.
Existe un alto porcentaje de casos que no cumplen todos los requisitos señalados. También se hallan otras formas de epilepsia de inicio clínico similar pero que no evolucionan de forma tan negativa.
Factores desencadenantes de las crisis en el síndrome de Dravet
Cuadro de fiebre
Cambios bruscos de la temperatura corporal (baño,…)
Determinados estímulos como patrones visuales, luces, etc…
Calor generado por el ejercicio físico
Emociones intensas
Los trastornos habituales asociados al espectro del Síndrome de Dravet
Coordinación, crecimiento y nutrición
Características del espectro autista y discapacidades de comunicación
Problemas cardiovasculares
Desarrollo y trastornos cognitivos
Salud dental
Disautonomía (problemas con las funciones corporales automáticas: regulación de la temperatura, disminución de la sudoración, función intestinal lenta y frecuencia cardíaca a veces rápida)
Infecciones respiratorias recurrentes e infecciones del oído
Condiciones ortopédicas (como deformidades de los pies, curvatura de la columna vertebral o escoliosis y dificultades para caminar)
Trastornos del sueño
SUDEP
La mortalidad asociada al Síndrome de Dravet ha sido descrita en estudios previos con porcentajes superiores al 15%, generalmente debida a SUDEP o muerte inesperada del paciente con epilepsia. Se ha producido una leve mejoría de los datos en los últimos años que puede ser debida a un mejor manejo de las crisis y a una mejor elección de los fármacos antiepilépticos.
|
Tipos de crísis
¿Qué son?
Las crisis o ataques epilépticos son episodios de alteración de la función del cerebro. Durante ellas la señal eléctrica que utilizan las neuronas para comunicarse entre sí se propaga de forma desordenada y excesiva a otras neuronas vecinas e incluso a regiones alejadas. Las crisis epilépticas afectan a la corteza cerebral, donde se albergan funciones cerebrales que requieren de nuestra voluntad (por ejemplo hablar, entender, pensar, memorizar, mover los músculos, prestar atención) y es la que recibe información de los órganos de la visión, el oído, el tacto, el olfato y el gusto. Cuando se produce una crisis epiléptica se alteran una o varias de estas funciones, por este motivo, los síntomas durante las crisis epilépticas son muy variables y reflejan la función de la región de corteza cerebral que ha provocado el inicio de la crisis y a la que se ha propagado.
Las crisis suelen tener una duración breve, menos de dos minutos, a veces sólo unos segundos. Inmediatamente después de una crisis las neuronas afectadas pierden transitoriamente su función, pues durante la excitación eléctrica consumen toda su energía. Por este motivo, después de cada crisis los enfermos suelen encontrarse cansados, somnolientos, confusos, con debilidad en alguna parte del cuerpo o con dificultad para hablar durante minutos o varias horas. Pasado este tiempo las neuronas recuperan la energía y reanudan su función normal.
¿Por qué se producen las convulsiones en el síndrome de Dravet?
¿Qué es un status epilépticus?
El status epilepticus o estatus epiléptico es una emergencia neurológica que requiere una atención inmediata. El diagnóstico y el tratamiento deben ser continuos a lo largo de los primeros minutos hasta su resolución. El estatus epiléptico puede ser convulsivo o no convulsivo y la monitorización electroencefalográfica continua es de gran ayuda para el diagnóstico y para valorar la respuesta al tratamiento. Las benzodiazepinas y la fenitoína son los fármacos de elección de primera y segunda línea. No existe consenso sobre los tratamientos de tercera y cuarta línea, entre los que se encuentran: fenobarbital, valproato, levetiracetam, propofol, midazolam, barbitúricos y otros.
Tipos de crisis
Las crisis epilepticas se dividen en dos grupos principales: crisis focales o parciales y crisis generalizadas. Las crisis focales se originan en una región circunscrita de la corteza cerebral, y desde ahí pueden propagarse a otras regiones. Si se asocian con confusión o alteración de la conciencia se denominan crisis parciales complejas, si no se asocian con alteración de la conciencia, son crisis parciales simples. Las crisis generalizadas se originan simultáneamente en los dos hemisferios cerebrales y se asocian con alteración de la conciencia.
Los tipos de convulsiones en el Síndrome de Dravet son:
Crisis tónico-clónicas. Son crisis generalizadas (implican los dos hemisferios cerebrales). Comienzan con una rigidez en brazos y piernas (fase tónica), luego le siguen movimientos espasmódicos en brazos, piernas y cabeza (fase clónica). Los afectados pierden la consciencia durante la convulsión. Estos episodios son habituales en el Síndrome de Dravet a cualquier edad. Pueden ser parciales, esto es, que sólo afectan a un lado del cuerpo.
- Mioclonías. Las mioclonías son movimientos involuntarios, breves, bruscos y similares a sacudidas que provocan una contracción muscular brusca. Suelen ser generalizadas, aunque también pueden ser focales, causando movimientos bruscos de cabeza, un brazo, o los párpados.
- Crisis parciales. Las crisis parciales afectan a un área limitada del cerebro y el afectado mantiene la consciencia. En el caso de una crisis parcial compleja se pierde dicha consciencia.
- Crisis de Ausencia. La mayoria de las ausencias típicas duran solo unos pocos segundos y con mayor frecuencia involucran episodios de mirada fija o "ausencias". Estos episodios pueden ocurrir muchas veces al dia. Las ausencias atípicas comienzan de manera más lenta y duran más.
El Síndrome de Dravet se considera un tipo de canalopatía dependiente de voltaje. Aproximadamente el 75% de los pacientes con esta enfermedad presenta una mutación puntual del gen SCN1A, que codifica para la subunidad α de tipo I del canal de Na+,Nav1.1, y lleva a la haploinsuficiencia del mismo (no se encuentran en la dosis génica adecuada). Explicado de una forma más sencilla de entender, el gen SCN1A tiene la información para construir una proteína que forma el canal de sodio en las neuronas, al producirse la mutación se origina una proteína con unas características funcionales alteradas.
Hasta la actualidad se han descrito más de 500 mutaciones diferentes en SCN1A en pacientes con Síndrome de Dravet, distribuidas aleatoriamente a lo largo de los 26 exones que componen el gen. La mayoría de las mutaciones de SCN1A presentes en casos con Síndrome de Dravet son de novo, es decir, que se expresa por primera vez en una familia. Pero existe un 5-10% de mutaciones Familiares.
Otros genes han sido implicados recientemente en el síndrome de Dravet: PCDH19, SCN9A SCN8A.
Aproximadamente un 5% de los casos diagnosticados como Síndrome de Dravet encuentran la causa molecular en la mutacion del gen PCDH19. Define una entidad clínica independiente de Síndrome de Dravet, la Epilepsia Limitada a Mujeres con Retraso Mental (EFMR), un desorden con un patrón de herencia ligado al cromosoma X poco habitual.
Otros genes, como SCN8A y SCN9A, han sido descritos como genes modificadores del fenotipo en síndrome y podrían explicar el amplio espectro fenotípico de los pacientes con síndrome de Dravet. Aproximadamente un 20% de los pacientes con diagnostico clínico sugestivo de Síndrome de Dravet no presenta mutación en los genes asociados a la enfermedad conocidos hasta ahora y resultan en un diagnostico genético molecular no caracterizado.
Representación esquemática de una neurona y la localizacion celular del canal de Na+ (SCN1A, SCN2A, SCN9A, SCN1B), canal de Cl- (GABRG2) y Protocadherina 19 (PCDH19).
Un diagnóstico genético positivo no implica que se vaya a desarrollar un Síndrome de Dravet completo. Hay otros factores que influyen en la enfermedad ya que hay otros genes que hacen de reguladores.
Aún hay mucho por comprender sobre las causas del síndrome de Dravet y la investigación se halla en una fase inicial.
¿Qué es una mutación?
La mutación es una modificación que se produce en el ADN de una persona. El ADN es una molécula de gran tamaño que guarda y transmite de generación en generación toda la información necesaria para el desarrollo de todas las funciones biológicas de un organismo. El ADN está formado por la unión paralela de dos cadenas, cada cadena se encuentra conformada por 4 diferentes nucleótidos. El ADN de todos los organismos vivos está formado por solo éstos cuatro nucleótidos.
El ADN es una acumulación de genes (fragmentos de ADN) y cada gen es la clave para la producción de una proteína. Por explicarlo de una forma sencilla, sería algo así como las instrucciones para construir un mueble.
El gen SCN1A contiene las instrucciones para construir los canales de sodio en las neuronas. Las neuronas con las células que están en el sistema nervioso, son células especiales ya que están diseñadas para transmitir información. La información la transmiten mediante la sinápsis. La sinapsis conecta unas neuronas con otras y transmite el impulso nervioso. Este impulso nervioso es un impulso eléctrico que permite desde mover un músculo hasta realizar los procesos cognitivos más complejos (ver, oír, hablar, tener conciencia, etc.). La mutación del gen provoca un problema en el sistema que regula los impulsos eléctricos. Es decir, la mutación provoca un cambio o error en las instrucciones para construir de forma correcta el mueble.
Tipos de mutaciones
Existen varios tipos de mutaciones. Para entender mejor los conceptos vamos a imaginarnos ese libro de instrucciones del que hemos hablado antes como un collar de perlas. Así el ADN sería como un collar de perlas Este collar tiene muchas perlas que básicamente puede ser de cuatro tipos (adenina - A, timina - T, citosina - C o guanina- G).
Aquí se pueden consultar la lista actualizada de las mutaciones halladas hasta la fecha http://www.molgen.ua.ac.be/SCN1AMutations/
|
Diagnóstico
En el primer año de vida, y especialmente en los primeros episodios, es frecuente confundir el Síndrome de Dravet con convulsiones febriles. La diferencia entre una y otra entidad es relevante, ya que en el caso de las convulsiones febriles no se trata de epilepsia, por lo que no está indicado un tratamiento crónico con fármacos antiepilépticos. El síndrome de Dravet, en cambio, precisa de medidas de prevención de la fiebre, cobertura antiepiléptica y tratamiento agresivo inicial ante la posibilidad de un estado epiléptico, así como la instauración de un programa de atención temprana, por lo que es importante que el paciente sea tratado adecuadamente y por un especialista con la menor demora posible.
Hasta hace relativamente poco, el diagnóstico sólo era posible hacia los 2-4 años de vida debido a la necesidad de esperar a la evolución para poder establecerlo, pero el desarrollo del test genético, el estudio molecular y el mejor conocimiento de la clínica pueden adelantarlo actualmente.
Aunque una primera crisis con fiebre en el síndrome de Dravet puede ser similar a una convulsión febril, hay, no obstante, algunas particularidades en el Dravet que permiten sospecharlo. Las crisis febriles del síndrome de Dravet aparecen en muchos casos antes de los 7 meses, tienden a ser prolongadas y a repetirse en un período breve. Además, suelen ser hemiclónicas, aunque las clónicas bilaterales o generalizadas tampoco son infrecuentes. La temperatura que las desencadena puede no ser excesivamente elevada.
Aspectos a tener en cuenta en un diagnóstico de Síndrome de Dravet y espectros asociados:
La importancia del Test Genético
El Síndrome de Dravet es una enfermedad para la que no se conoce cura ni fármacos específicos y que no solo es refractaria a tratamiento con los antiepilépticos disponibles actualmente, sino que estos pueden producir síntomas más severos y de difícil control. Así, se hace imprescindible un diagnostico precoz que combine diagnóstico clínico y genético.
El diagnóstico clínico se realiza a través de una detallada historia de crisis convulsivas, tipo de convulsión, edad de debut, etc., además de pruebas diagnosticas como electroencefalogramas y tomografías computerizadas. El diagnostico genético de calidad,como el ofrecido de forma gratuita por la Fundación, permite identificar la causa genética molecular del síndrome de Dravet. De esta forma, los pacientes y sus familiares se podrán beneficiar de un apropiado consejo genético familiar y de un servicio de cribado prenatal si desean tener más descendencia. Por otra parte, permite confirmar el diagnóstico clínico previo, ayudando a la administración de un correcto tratamiento para estos pacientes y un mejor seguimiento de la enfermedad.
Por todo ello, la inclusión del diagnóstico genético en la práctica clínica habitual, en combinación con el diagnostico clínico proporcionado por los neurólogos, permitirá establecer una rutina diagnóstica de calidad que beneficiara a los pacientes con SD.
Tratamiento y evolución
La rareza de la enfermedad y su identificación, relativamente reciente, hace que el diagnóstico a largo plazo y la esperanza de vida constituyan una incógnita a día de hoy. Fuente de información: http://www.dravetfoundation.eu/es/sobre-dravet/que-es |

No hay comentarios:
Publicar un comentario